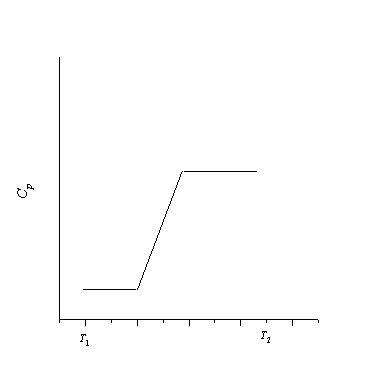Wzór na obliczanie entropii. Entropia
Jeżeli układ termodynamiczny przeszedł z jednego stanu do drugiego, to ilość odbieranego przez niego ciepła zależy nie tylko od stanu początkowego i końcowego, ale także od rodzaju procesu przejścia. Innymi słowy, ilość ciepła jest funkcją procesu, a nie stanu. W poszukiwaniu funkcji stanu rozważmy wielkość, której przyrost jest równy stosunkowi ciepła pochłoniętego na odcinku procesu odwracalnego do temperatury:
Ta wartość nazywana jest entropią lub zmniejszoną ilością ciepła. Pojęcie entropii wprowadził w 1865 r. R. Clausius. Łatwo pokazać, że suma wszystkich przyrostów entropii w cyklu Carnota jest równa zeru. Rzeczywiście, dla procesów adiabatycznych 2-3 i 4-1 dS= Och, ponieważ dQ = 0 (ryc. 13.3). A dla pozostałych procesów izotermicznych 1-2 i 3^1 całkowanie (13.12) uwzględniając (13.5) i (13.7) daje
Wynik ten jest ważny niezależnie od wyboru płynu roboczego. Można wykazać, że zmiana entropii wynosi zero nie tylko w cyklu Carnota, ale także w każdym innym cyklu odwracalnym. Zatem entropia jest funkcją stanu, a jej wartości na początku i na końcu procesu kołowego są takie same.
Można wykazać, że entropia układu wykonującego nieodwracalny cykl wzrasta. Ogólnie rzecz biorąc, to prawda Nierówność Clausiusa: entropia układu zamkniętego wzrasta (w przypadku procesów nieodwracalnych) lub pozostaje stała (w przypadku procesów odwracalnych)
Znajdźmy teraz za pomocą (12.9) i (12.3) zmianę entropii w procesach gazu doskonałego:
Tutaj ciśnienie jest wyrażone za pomocą równania Mendelejewa-Clapeyrona. Całkowanie wynikowego wyrażenia daje
Należy podkreślić, że o zmianie entropii decydują tylko stany początkowe i końcowe gazu doskonałego i nie zależy ona od charakteru procesu przejścia. Ze wzoru (13.17) wynika, że w procesie izotermicznym
oraz w procesie izochorycznym
Jak już wspomniano, z definicji entropii wynika, że dla procesu adiabatycznego dS= 0. Entropia jest mierzona w dżulach na kelwin (J/K). Entropia ma właściwość addytywności: entropia układu jest równa sumie entropii ciał wchodzących w skład układu. Wynika to z faktu, że ciepło pochłaniane przez układ składa się z porcji ciepła pochłanianego przez jego części. Własność addytywności posiada również energia wewnętrzna, masa, objętość (a np. temperatura i ciśnienie takiej własności nie posiadają).
Entropia jest nie tylko pojęciem termodynamicznym, ale także statystycznym. Jest to związane z termodynamicznym prawdopodobieństwem stanu układu. Prawdopodobieństwo termodynamiczne W stanu układu- jest to liczba sposobów realizacji danego stanu układu makroskopowego lub liczba mikrostanów realizujących dany makrostan. Termodynamiczne prawdopodobieństwo stanu układu składającego się tylko z 10 cząsteczek gazu wynosi około 1000, aw rzeczywistych układach cząsteczek jest o wiele rzędów wielkości większe. Dlatego okazało się, że wygodniej jest używać percepcji w termodynamice, a nie ilości W, i jego logarytm w W. Temu ostatniemu można nadać wymiar (J/K) mnożąc przez stałą Boltzmanna do. Entropia układu i prawdopodobieństwo termodynamiczne są ze sobą powiązane Formuła Boltzmanna: gdzie do jest stałą Boltzmanna.
Zatem entropia jest równa logarytmowi liczby mikrostanów, z którymi dany makrostan może zostać zrealizowany. Im większa liczba mikrostanów realizujących dany makrostan, tym większa entropia. W stanie równowagi - najbardziej prawdopodobnym stanie układu - liczba mikrostanów jest maksymalna, entropia też jest maksymalna.
Entropia jest często uważana za miarę nieuporządkowania w systemie. Wynika to z faktu, że systemy uporządkowane mają zwykle znacznie mniej mikrostanów niż układy nieuporządkowane. Tak więc 100 molekuł w dwóch połówkach naczynia może być rozmieszczonych równo, zgodnie z teorią prawdopodobieństwa, na ogromną liczbę sposobów. A umieszczenie ich wszystkich na jednej połowie odpowiada tylko dwóm opcjom. Dlatego cząsteczki mają tendencję do rozkładu w przybliżeniu równomiernie w objętości, a jednocześnie entropia jest maksymalna. Rozważmy na przykład rozkład cząsteczek gazu doskonałego. W przypadku gazu doskonałego najbardziej prawdopodobnym stanem odpowiadającym maksymalnej entropii będzie równomierny rozkład cząsteczek. Jednocześnie realizowany jest maksymalny „bałagan”, ponieważ możliwości konfiguracji będą maksymalne. Prawdziwe procesy nieodwracalne w układzie zamkniętym prowadzą do wzrostu jego entropii – to zasada zwiększania entropii.
Należy podkreślić, że powyższe twierdzenia, w tym druga zasada termodynamiki, mają charakter statystyczny i mogą nie mieć zastosowania dla układów o małej liczbie cząstek.
WYKŁAD 11
Diagramy fazowe
faza nazywa się stan skupienia materii, charakteryzujący się tym, że zajmuje on określony obszar przestrzeni, aw obrębie tego obszaru parametry i właściwości substancji albo pozostają stałe, albo zmieniają się w sposób ciągły. Ten obszar przestrzenny jest oddzielony od innych części przestrzeni granicą. Masa substancji zawartej w jednej fazie może zmieniać się w czasie. W tym przypadku mówi się o przejście fazowe. Przejście fazowe odbywa się przez granicę faz. Istnieją następujące najczęstsze przejścia fazowe:
gotowanie (przejście substancji z cieczy w parę);
kondensacja (przejście substancji z pary w ciecz);
krystalizacja, twardnienie (przejście substancji ze stanu ciekłego w stan stały);
topnienie (przejście substancji ze stanu stałego w ciekły).
Fazy są wygodnie przedstawione na diagramach fazowych. Diagram fazowy to płaszczyzna z wpisanym na nią kartezjańskim układem współrzędnych, wzdłuż osi których wykreślone są wartości pary podstawowych parametrów termodynamicznych. Ta płaszczyzna jest podzielona na kilka regionów, z których każdy reprezentuje określoną fazę. Diagram fazowy przedstawia również główne izolinie (linie stałości głównych parametrów termodynamicznych: izochory, izobary, izotermy, izentropy, izoetapy oraz linie stałej suchości).
Najczęściej spotykane są diagramy fazowe postaci T-S, P-V, H-S, H-lgP. Rozważ diagram faz T-S. na ryc. 31 pokazuje główne fazy i granice faz:
 |
F - ciecz
W + T - ciecz + ciało
NK - region nadkrytyczny
G - obszar gazowy; VP - mokra para
bkc to krzywa nasycenia. Charakteryzuje nasycony stan substancji.
bk to linia cieczy nasyconej. nasycona ciecz- jest to ciekły stan skupienia, charakteryzujący się tym, że dostarczenie dowolnie małej ilości ciepła prowadzi do intensywnego tworzenia się pary wodnej.
kc - linia sucha para nasycona. Jest to gazowy stan skupienia, charakteryzujący się tym, że dowolnie małe ochłodzenie prowadzi do zapoczątkowania procesu kondensacji.
abc - linia potrójnych punktów. potrójny punkt- jest to stan skupienia charakteryzujący się równowagowym współistnieniem jednocześnie trzech faz: stałej, ciekłej i gazowej. Równowaga fazowa charakteryzuje się tym, że nie ma przejścia fazowego między fazami. W stałych warunkach zewnętrznych równowaga faz może współistnieć w nieskończoność. Aby dwie fazy znajdowały się w stanie równowagi, muszą być spełnione trzy warunki: 1) fazy muszą mieć takie samo ciśnienie; 2) fazy muszą mieć tę samą temperaturę; 3) fazy muszą mieć potencjał chemiczny.
be - linia początku procesu krzepnięcia lub końca procesu topienia.
ad - linia końca procesu krzepnięcia lub początku procesu topienia.
dek to krytyczna izoterma temperatury.
P=P cr - izobara krytyczna.
k- punkt krytyczny. Charakteryzuje się tym, że w temperaturze powyżej krytycznej niemożliwe jest uzyskanie cieczy metodą kompresji izotermicznej. Krytyczne ciśnienie i temperatura to ciśnienia i temperatury poniżej punktu krytycznego.
Region G - strefa gazowa. W tym regionie panuje ciśnienie poniżej krytycznego i temperatura powyżej krytycznego. Obszar gazowy charakteryzuje się tym, że stan gazu w tym obszarze jest opisany równaniem stanu gazu doskonałego.
obszar PP - obszar pary przegrzanej. Znajduje się w temperaturze poniżej temperatury krytycznej i na prawo od linii kc. Obszar ten charakteryzuje się tym, że zachowanie się w nim materii opisuje równanie van der Waalsa lub zmodyfikowane równanie gazu doskonałego
| , | (130) |
gdzie z jest współczynnikiem ściśliwości (współczynnikiem korygującym uwzględniającym odchylenie zachowania substancji rzeczywistych od gazu doskonałego).
Region W+R - strefa mokrej pary. Ograniczona przez krzywą nasycenia i linię punktu potrójnego. Jest to obszar dwufazowy charakteryzujący się stanem równowagi nasyconej pary i nasyconej cieczy. Jest to obszar procesów kondensacji i wrzenia.
Region J. - obszar przechłodzonej cieczy. Ogranicza ją od góry izoterma krytyczna, z prawej strony linia cieczy nasyconej, z lewej linia początku krystalizacji.
Obszar T+L jest dwufazowym obszarem równowagowego współistnienia fazy ciekłej i stałej. Ten obszar procesów krzepnięcia (krystalizacji) topienia.
Obszar T+P to dwufazowy obszar równowagowego współistnienia pary nasyconej i ciała stałego. Z góry obszar ten jest ograniczony linią potrójnych punktów. potrójny punkt nazywa się stanem równowagi trzech stanów agregacji jednocześnie. Jest to obszar procesów sublimacji i desublimacji. Sublimacja Nazywa się proces przejścia z fazy stałej w fazę gazową. desublimacja zwany procesem przejścia pary nasyconej w fazę stałą.
Obszar NC jest obszarem nadkrytycznego stanu materii. Znajduje się w ciśnieniu i temperaturze powyżej krytycznej. Charakteryzuje się tym, że substancja w tym stanie ma właściwości zarówno cieczy, jak i gazu.
na ryc. 32 przedstawia główne linie technologiczne.
Liniami ciągłymi zaznaczono izobary odpowiadające ciśnieniom Р 1 , Р 2 , Р 3 = Р cr i Р 4 . W tym przypadku między tymi ciśnieniami relacje Р 1<Р 2 <Р 3 <Р 4 . Следует отметить, что процессы, что процессы, протекающие в двухфазных областях, изображаются горизонтальными линиями, т.е. эти изобарные процессы одновременно являются изотермическими. Изобара с давлением Р 4 лежит выше критической точки не проходит через область влажного пара, а сразу из области надкритического состояния попадает в область переохлаждённой жидкости. Изобара с давлением Р 1 лежит ниже линии тройных точек, также не проходит через область влажного пара, а из области перегретого пара попадает в область твёрдого состояния вещества посредством процесса десублимации. Изобара с давлением Р 3 касается критической точки. Изобара с давлением Р 2 , проходя через область влажного пара, реализует процесс кипения или конденсации.
Izochory w v 1 i v 2 (v 1 > v 2) przedstawione liniami przerywanymi leżą na diagramie T-S bardziej stromo niż izobary. Należy zauważyć, że izochory w regionach dwufazowych nie pokrywają się z izotermami; nie poziome.
Isenthalps h 1 , h 2 i h 3 (h 1 > h 2 > h 3) są pokazane liniami przerywanymi. Można zwrócić uwagę na fakt, że wraz ze spadkiem temperatury zwiększa się kąt nachylenia izentalpe do osi S.
Mokra para
Wilgotna para to stan skupienia, w którym para nasycona i nasycona ciecz znajdują się w równowadze. Równowaga wynika z równości ich temperatur i ciśnień. Obszar mokrej pary znajduje największe zastosowanie w urządzeniach ciepłowniczych i niskotemperaturowych, ponieważ. w tym obszarze najłatwiej jest realizować procesy istotne w zastosowaniach technicznych (izoterma).
Obszar mokrej pary przedstawiony na schemacie T-S pokazano na ryc. 33.
 |
Punkt „a” charakteryzuje stan mokrej pary, w którym nasycona ciecz i nasycona para znajdują się w równowadze w określonych ułamkach masowych.
Para nasycona jest w stanie , a stan cieczy nasyconej charakteryzuje punkt . Niech mokra para a w stanie punktowym a zajmuje pewną objętość , gdzie m jest masą mokrej pary; v a - określona objętość mokrej pary. Tę samą objętość można uznać za sumę objętości nasyconej cieczy i nasyconej pary
| , | (131) |
gdzie jest objętość nasyconej cieczy;
Objętość pary nasyconej;
Masa nasyconej cieczy;
Masa pary nasyconej;
Objętość właściwa cieczy nasyconej w stanie skupienia;
Specyficzna objętość pary nasyconej w stanie punktowym.
W tym przypadku relacja
Dzieląc obie części równania przez m w ostatnim wyrażeniu, otrzymujemy równanie wyrażające objętość właściwą pary mokrej w przeliczeniu na objętości właściwe cieczy nasyconej i pary nasyconej
| . | (133) |
W tym wyrażeniu stopień suchości pary mokrej, który pokazuje udział masowy pary nasyconej w parze mokrej. Jeśli x=1, to para mokra składa się wyłącznie z pary nasyconej. Jeśli x=0, to mokra para składa się wyłącznie z nasyconej cieczy. Stopień wysuszenia może przyjmować dowolną wartość z przedziału od 0 do 1. Zbiór wszystkich punktów obszaru pary mokrej na wykresie T-S, które mają jedną wartość stopnia wysuszenia, nazywamy liniami stałej suchości (patrz rys. 33).
Dokładnie w ten sam sposób rozumowania, wykorzystując właściwość addytywności entalpii i entropii, można otrzymać wyrażenia
| , | (134) |
| , | (135) |
gdzie jest entalpią właściwą cieczy nasyconej w stanie punktowym;
Entalpia właściwa pary nasyconej w stanie punktowym;
Entropia właściwa cieczy nasyconej w stanie punktowym;
Entropia właściwa pary nasyconej w stanie punktowym.
Wyraź z ostatniego równania x
| . | (136) |
Ze wzoru tego wynika, że aby zwiększyć stopień wysuszenia, należy zwiększyć entropię, tj. zastosować ciepło do mokrej pary. W takim przypadku udział nasyconej cieczy zmniejszy się, a udział nasyconej pary wzrośnie. Parametry cieczy nasyconej i pary nasyconej nie ulegną w tym przypadku zmianie. Ten proces nazywa się gotowaniem. Jeżeli ciepło zostanie usunięte z mokrej pary, wówczas entropia zmniejszy się, co oznacza, że zmniejszy się stopień wysuszenia, tj. substancja przejdzie ze stanu nasyconej pary do stanu nasyconej cieczy. Ten proces nazywa się kondensacją.
Aby całkowicie zamienić 1 kg nasyconej cieczy w stan suchej nasyconej pary, konieczne jest dostarczenie określonej ilości ciepła, która nazywa się ciepłem właściwym parowania r, .
W procesie izobarycznym, czyli wrzeniu lub skraplaniu, ciepło dostarczane lub usuwane jest równe zmianie entalpii. Dlatego relacja
WYKŁAD 12
Cykl termodynamiczny
cykl termodynamiczny nazywa się zamkniętym procesem termodynamicznym, tj. proces, w wyniku którego system powraca do swojego pierwotnego stanu. Inną definicję cyklu termodynamicznego można podać jako sekwencję procesów termodynamicznych, których wykonanie doprowadza układ do stanu początkowego. Piszemy pierwszą zasadę termodynamiki dla układu zamkniętego w postaci
Ponieważ system powraca do swojego pierwotnego stanu, to . Wynikiem jest uogólnione równanie cyklu termodynamicznego
gdzie Q 1 to całkowite ciepło dostarczane w cyklu do systemu;
Q 2 to całkowite ciepło usunięte z układu w cyklu.
Podstawiając (140) do (139) otrzymujemy
| . | (141) |
W tym wyrażeniu odebrane ciepło przyjmuje się jako dodatnie, ponieważ znak usuniętego ciepła jest brany pod uwagę we wzorze z minusem przed Q 2.
Równanie (141) umożliwia podział obiegów termodynamicznych na dwa typy:
1. jeśli , to cykl nazywamy prostym;
2. jeśli , to cykl jest również nazywany odwrotnością.
cykl bezpośredni
cykl bezpośredni zwaną także mocą cieplną. Jest to cykl, w wyniku którego system wytwarza, tj. wykonuje pracę z powodu ciepła dostarczonego do układu.
Schemat ideowy urządzenia realizującego cykl mocy bezpośredniej lub termicznej pokazano na ryc. 34.
Na tym zdjęciu:
TDS(M) to układ termodynamiczny (maszyna), który wykonuje cykl;
HI to gorące źródło o temperaturze T HI. Jest rozumiany jako zbiór ciał środowiskowych, które przekazują ciepło Q 1 do układu termodynamicznego.
CI - źródło zimna lub lodówka o temperaturze T CI. Jest to zbiór ciał środowiskowych, którym układ, wykonując cykl, oddaje ciepło Q 2 . Aby obwód pokazany na rys. 34, źródło zimna musi mieć temperaturę T HI niższą niż temperatura źródła gorącego T HI (T HI<Т ГИ). Кроме того, температура холодного источника должна быть меньше минимальной температуры системы в цикле, а температура горячего источника должна быть больше максимальной температуры системы.
 |  |
Ryż. 35. Ryc. 36.
na ryc. 35 pokazuje cykl mocy cieplnej na wykresie T-S. Procesowi 1a2 towarzyszy dopływ ciepła Q 1 , ponieważ entropia wzrasta. W tym przypadku dostarczone ciepło jest równe polu powierzchni pod linią 1a2. W procesie 2b1 ciepło Q 2 jest usuwane, ponieważ entropia maleje, a ciepło to jest równe polu powierzchni pod linią 2b1. Z rysunku widać, że pole figury m1a2n jest większe niż pole m1b2n, a więc Q 1 > Q 2 , a cykl ten jest prosty. W rezultacie różnica między ciepłem dostarczonym a odprowadzonym jest równa pracy cyklu i jest równa powierzchni cyklu.
na ryc. 36 pokazuje cykl mocy cieplnej na wykresie P-V. Procesowi 1a2 towarzyszy wykonanie pracy L 1 a 2 , ponieważ objętość wzrasta w tym procesie. W tym przypadku wykonana praca jest równa polu pod linią 1a2. W procesie 2b1 praca L 2 b 1 jest wydawana, ponieważ objętość maleje, a praca ta jest równa polu pod linią 2b1. Z rysunku widać, że pole figury m1a2n jest większe niż pole m1b2n, a więc L 1 a 2 > L 2 b 1 , a cykl ten jest prosty. W rezultacie różnica między pracą wykonaną a pracą wydatkowaną jest równa pracy cyklu i jest równa polu ograniczonemu przez cykl.
Każdy cykl, zarówno bezpośredni, jak i odwrotny, charakteryzuje współczynnik sprawności, który ocenia efektywność procesu konwersji energii.
Ponieważ, zgodnie z definicją cyklu bezpośredniego, to sprawność jest zawsze mniejsza od jedności. Proces zamiany energii cieplnej na pracę użyteczną w cyklu jest tym bardziej efektywny, im wartość sprawności cyklu jest bliższa jedności.
cykle odwrotne
Cykl odwrotny to cykl, w którym doprowadzone ciepło jest mniejsze niż ciepło odebrane. W rezultacie praca cyklu odwrotnego jest ujemna, tj. wymaga pracy.
Schemat ideowy urządzenia realizującego cykl odwrotny pokazano na ryc. 37.
 |  |
Ryż. 38. Ryc. 39.
na ryc. 38 pokazuje cykl odwrotny na schemacie T-S. Procesowi 1a2 towarzyszy dopływ ciepła Q 1 , ponieważ entropia wzrasta. W tym przypadku dostarczone ciepło jest równe polu powierzchni pod linią 1a2. W procesie 2b1 ciepło Q 2 jest usuwane, ponieważ entropia maleje, a ciepło to jest równe polu powierzchni pod linią 2b1. Z rysunku widać, że obszar figury m1a2n jest mniejszy niż obszar m1b2n, dlatego Q 1 na ryc. 39 pokazuje odwrotny cykl na wykresie P-V. Procesowi 1a2 towarzyszy wykonanie pracy L 1 a 2 , ponieważ objętość wzrasta w tym procesie. W tym przypadku wykonana praca jest równa polu pod linią 1a2. W procesie 2b1 praca L 2 b 1 jest wydawana, ponieważ objętość maleje, a praca ta jest równa polu pod linią 2b1. Z rysunku widać, że obszar figury m1a2n jest mniejszy niż obszar m1b2n, dlatego L 1 a 2 Odwrotne cykle termodynamiczne dzielą się na trzy typy: 1. cykle chłodzenia; 2. cykle pompy ciepła; 3. cykle łączone. Cykl chłodzenia pokazano na ryc. 40 pod cyfrą rzymską I. Jest to obieg odwrotny, w którym wydatkowana jest praca w celu odebrania ciepła Q 1 z chłodzonego obiektu w temperaturze T 00 poniżej temperatury otoczenia T OC. Cykle chłodnicze realizowane są w instalacjach niskotemperaturowych, w szczególności w domowych lodówkach. W tym przypadku ciepło Q 1 dostarczane do substancji roboczej (freonu) jest ciepłem usuwanym z produktów w zamrażarce. Obieg II pompy ciepła jest obiegiem odwrotnym, w którym praca jest wydatkowana na dostarczenie ciepła Q 2 do ogrzewanego obiektu o temperaturze T H O wyższej niż temperatura otoczenia T OS. Cykl ten realizowany jest przez domowe klimatyzatory pracujące w trybie ogrzewania pomieszczeń. Ogrzewanym obiektem jest w tym przypadku powietrze pokojowe. Temperatura ogrzewanego obiektu jest temperaturą pokojową. Powietrze zewnętrzne o niskiej temperaturze działa jak środowisko. Ciepło Q 2, które idzie w tym przypadku do ogrzania pomieszczenia i jest określone wyrażeniem (144), jest większe niż ciepło, które zostałoby dostarczone, gdyby pomieszczenie było ogrzewane grzejnikiem elektrycznym, w którym energia elektryczna L jest zamieniana na ciepło energia. Obieg kombinowany III jest obiegiem odwrotnym, w którym praca jest wydatkowana na usunięcie ciepła Q 1 z chłodzonego obiektu znajdującego się w temperaturze T 00 poniżej temperatury otoczenia i jednoczesne dostarczenie ciepła Q 2 do ogrzewanego obiektu znajdującego się w temperaturze THO powyżej temperatury otoczenia temperatura. Urządzeniem realizującym cykl łączony jest lodówka domowa zlokalizowana w dzielnicy mieszkalnej. Z kolei z zewnątrz tego pomieszczenia napływa powietrze o niskiej temperaturze. W tym przypadku przedmiotem ogrzewania, do którego dostarczane jest ciepło Q 2 (usunięte z obiegu), jest powietrze w pomieszczeniu o temperaturze pokojowej. Przedmiotem chłodzenia są produkty znajdujące się w zamrażarce, z których ciepło Q1 jest odprowadzane i dostarczane do krążącego w lodówce freonu. Współczynnik wydajności cyklu chłodzenia nazywany jest współczynnikiem chłodzenia ε. Energią użyteczną jest w tym przypadku ciepło Q 1 odebrane z chłodzonego obiektu i dostarczone do substancji roboczej, która wykonuje obieg. Zużyta energia to praca wejściowa L. Dlatego Z wyrażenia tego widać, że współczynnik ogrzewania jest zawsze większy od jedności, a obieg pompy ciepła jest tym wydajniejszy, im większa jest wartość μ powyżej jedności. Współczynnik sprawności cyklu łączonego nie ma specjalnej nazwy i jest oznaczony jako k. Energią użyteczną jest w tym przypadku ciepło Q 1 odebrane z chłodzonego obiektu i jednocześnie ciepło Q 2 dostarczone do ogrzewanego obiektu. Zużyta energia to praca wejściowa L. Dlatego Z tego wyrażenia widać, że współczynnik sprawności cyklu łączonego jest oczywiście większy niż jeden, WYKŁAD 13 Odwracalny cykl Carnota Wszystkie cykle, zarówno bezpośrednie, jak i odwrotne, dzielą się na 2 typy: odwracalny i nieodwracalny. cykl odwracalny Nazywa się cykl składający się tylko z procesów odwracalnych. Nieodwracalny cykl to cykl, w którym występuje co najmniej jeden proces nieodwracalny. Aby proces był odwracalny, musi być w równowadze, tj. musi płynąć z nieskończenie małą prędkością. Jest to możliwe tylko wtedy, gdy różnica potencjałów oddziałujących z systemem i otoczeniem jest nieskończenie mała. Dla układu termodynamicznego oznacza to, że w przypadku odwracalnej wymiany ciepła z otoczeniem różnica temperatur między układem a otoczeniem musi być nieskończenie mała, tj. nie może występować opór cieplny między systemem a otoczeniem. Odwracalne rozszerzanie i kurczenie jest możliwe w przypadku nieskończenie małej różnicy ciśnień między układem a otoczeniem. Jest to możliwe tylko wtedy, gdy w układzie nie ma tarcia. Wynika z tego, że w układzie termomechanicznym istnieją dwa źródła nieodwracalności: 1. obecność oporu cieplnego między różnymi częściami układu, co prowadzi do skończonej różnicy temperatur podczas wymiany ciepła; 2. obecność tarcia w układzie (lub między układem a otoczeniem), które prowadzi do skończonej różnicy ciśnień. Spośród wszystkich obiegów termodynamicznych odwracalny cykl Carnota (bezpośredni) wyróżnia się na tej podstawie, że dla danej różnicy temperatur między źródłami gorącymi i zimnymi odwracalny cykl Carnota ma maksymalną możliwą wydajność. Odwracalny cykl Carnota pokazany na ryc. 41 i ryc. 42 składa się z dwóch adiabatów i dwóch izoterm. Ryż. 41. Ryc. 42 1-2 - proces ekspansji adiabatycznej. W tym procesie wykonywana jest praca L 12. 2-3 - proces kompresji izotermicznej. W tym procesie praca L 23 jest zużywana, a ciepło Q 23 jest usuwane. 3-4 - proces kompresji adiabatycznej. Proces ten pochłania pracę L 34 . 4-1 - proces rozszerzania izotermicznego. W tym procesie wykonywana jest praca L 41 i dostarczane jest ciepło Q 41. Głównymi procesami cyklu są procesy 4-1 i 1-2. Wykonują pracę cyklu. Pozostałe procesy mają charakter pomocniczy i mają na celu przywrócenie układu do stanu pierwotnego 4 przy jak najmniejszym nakładzie energii. Wyznaczmy efektywność odwracalnego cyklu Carnota η bcc: Z definicji efektywności (143) Podstawiając te wyrażenia do (148) i usuwając różnicę entropii, otrzymujemy Opierając się na tych samych rozważaniach, otrzymujemy Pokazuje to ta formuła Sprawność odwracalnego cyklu Carnota nie zależy od właściwości płynu roboczego wykonującego cykl Carnota i zależy tylko od temperatur gorących i zimnych źródeł
. Ten wniosek jest sformułowaniem pierwsze twierdzenie Carnota. §6 Entropia Zwykle każdy proces, w którym układ przechodzi z jednego stanu do drugiego, przebiega w taki sposób, że niemożliwe jest przeprowadzenie tego procesu w przeciwnym kierunku, tak aby układ przechodził przez te same stany pośrednie bez żadnych zmian w otaczających go ciałach. Wynika to z faktu, że część energii jest rozpraszana w procesie, na przykład w wyniku tarcia, promieniowania itp. Prawie wszystkie procesy w przyrodzie są nieodwracalne. W każdym procesie część energii jest tracona. Aby scharakteryzować rozpraszanie energii, wprowadzono pojęcie entropii. ( Wartość entropii charakteryzuje stan termiczny układu i określa prawdopodobieństwo zajścia w ten stan ciała. Im bardziej prawdopodobny dany stan, tym większa entropia. Wszystkim naturalnym procesom towarzyszy wzrost entropii. Entropia pozostaje stała tylko w przypadku wyidealizowanego procesu odwracalnego zachodzącego w układzie zamkniętym, czyli w układzie, w którym nie zachodzi wymiana energii z ciałami zewnętrznymi względem tego układu. Entropia i jej termodynamiczne znaczenie: Entropia- jest to taka funkcja stanu układu, której nieskończenie mała zmiana w procesie odwracalnym jest równa stosunkowi nieskończenie małej ilości ciepła wprowadzonego w tym procesie do temperatury, w której zostało wprowadzone. W końcowym procesie odwracalnym zmianę entropii można obliczyć za pomocą wzoru: gdzie całka jest pobierana ze stanu początkowego 1 systemu do stanu końcowego 2. Ponieważ entropia jest funkcją stanu, to własność całkijest jej niezależność od kształtu konturu (ścieżki), wzdłuż którego jest obliczana, dlatego całka jest określona tylko przez stan początkowy i końcowy układu. (1)
Δ
S> 0 (2)
Wyrażenia (1) i (2) dotyczą tylko układów zamkniętych, ale jeżeli układ wymienia ciepło z otoczeniem zewnętrznym, to jegoSmoże zachowywać się w dowolny sposób. Relacje (1) i (2) można przedstawić jako nierówność Clausiusa ∆S ≥ 0 tych. entropia układu zamkniętego może albo rosnąć (w przypadku procesów nieodwracalnych), albo pozostać stała (w przypadku procesów odwracalnych). Jeśli system dokonuje przejścia równowagi ze stanu 1 do stanu 2, wówczas zmienia się entropia gdzie dU oraz δAnapisany dla konkretnego procesu. Zgodnie z tym wzorem ΔSjest wyznaczany z dokładnością do stałej addytywnej. To nie sama entropia ma fizyczne znaczenie, ale różnica entropii. Znajdźmy zmianę entropii w procesach gazu doskonałego. tych. zmiany entropiiS Δ
S 1→2
gazu doskonałego podczas jego przejścia ze stanu 1 do stanu 2 nie zależy od rodzaju procesu. Dlatego dla procesu adiabatycznego δQ
= 0, następnie ∆ S= 0 =>
S= stała , to znaczy adiabatyczny proces odwracalny przebiega przy stałej entropii. Dlatego nazywa się to izentropem. W procesie izotermicznym (T= stała ; T 1
=
T 2
:
)
W procesie izochorycznym (V= stała ; V 1
=
V 2
;
)
Entropia ma właściwość addytywności: entropia układu jest równa sumie entropii ciał wchodzących w skład układu.S
=
S 1
+
S 2
+
S 3
+
...
Jakościową różnicą między ruchem termicznym cząsteczek a innymi formami ruchu jest jego losowość, nieuporządkowanie. Dlatego, aby scharakteryzować ruch termiczny, konieczne jest wprowadzenie ilościowej miary stopnia nieuporządkowania molekularnego. Jeśli weźmiemy pod uwagę dowolny stan makroskopowy ciała o pewnych średnich wartościach parametrów, to jest to coś innego niż ciągła zmiana bliskich mikrostanów, które różnią się od siebie rozmieszczeniem cząsteczek w różnych częściach objętości i w energia rozłożona między cząsteczkami. Liczba tych stale zmieniających się mikrostanów charakteryzuje stopień zaburzenia stanu makroskopowego całego układu,wnazywa się prawdopodobieństwem termodynamicznym danego mikrostanu. Prawdopodobieństwo termodynamicznewStany systemu to liczba sposobów, na jakie można zrealizować dany stan systemu makroskopowego, lub liczba mikrostanów, które realizują dany mikrostan (w≥ 1 i prawdopodobieństwo matematyczne ≤ 1
).
Zgodziliśmy się przyjąć logarytm jego prawdopodobieństwa ze znakiem minus jako miarę nieoczekiwanego zdarzenia: nieoczekiwanie stanu jest równe =-
Według Boltzmanna entropiaSukłady i prawdopodobieństwo termodynamiczne są powiązane w następujący sposób: gdzie - stała Boltzmanna ( Dlatego rzeczywiste procesy są nieodwracalne, to można argumentować, że wszystkie procesy w układzie zamkniętym prowadzą do wzrostu jego entropii – zasada rosnącej entropii. W statystycznej interpretacji entropii oznacza to, że procesy w układzie zamkniętym idą w kierunku zwiększania liczby mikrostanów, czyli od stanów mniej prawdopodobnych do bardziej prawdopodobnych, aż prawdopodobieństwo stanu osiągnie maksimum. §7 Druga zasada termodynamiki
Pierwsza zasada termodynamiki, wyrażająca zasadę zachowania energii i przemiany energii, nie pozwala na ustalenie kierunku przebiegu procesów t/d. Ponadto można sobie wyobrazić zestaw procesów, które nie są ze sobą sprzeczneIpoczątek m / d, w którym energia jest magazynowana, ale w naturze nie są realizowane. Możliwe sformułowania drugiego początku t/d: 1) prawo wzrostu entropii układu zamkniętego podczas procesów nieodwracalnych: każdy proces nieodwracalny w układzie zamkniętym zachodzi w taki sposób, że entropia układu wzrasta ΔS≥ 0 (proces nieodwracalny) 2) ΔS≥ 0 (S= 0 dla odwracalnego i ΔS≥ 0 dla procesu nieodwracalnego) W procesach zachodzących w układzie zamkniętym entropia nie maleje. 2) Ze wzoru Boltzmanna S = , zatem wzrost entropii oznacza przejście układu ze stanu mniej prawdopodobnego do stanu bardziej prawdopodobnego. 3) Według Kelvina: nie jest możliwy proces okrężny, którego jedynym rezultatem jest zamiana ciepła odbieranego z grzałki na równoważną mu pracę. 4) Według Clausiusa: nie jest możliwy proces okrężny, którego jedynym skutkiem jest przenoszenie ciepła z ciała mniej ogrzanego do ciała ogrzanego. Aby opisać układy t/d w 0 K, stosuje się twierdzenie Nernsta-Plancka (trzecie prawo t/d): entropia wszystkich ciał w równowadze dąży do zera, gdy temperatura zbliża się do 0 K Z twierdzenia Nernst-Planck podąża za tymC p= C v = 0 w 0 Do
§8 Maszyny termiczne i chłodnicze.
Cykl Carnota i jego efektywność
Ze sformułowania drugiego prawa t / d według Kelvina wynika, że perpetuum mobile drugiego rodzaju jest niemożliwe. (Maszyna perpetuum mobile to okresowo działający silnik, który działa poprzez chłodzenie jednego źródła ciepła). Zasada działania silnika cieplnego: z termostatu o temperaturze T 1 - grzałka, ilość ciepła jest odbierana na cyklQ 1
i termostat z temperaturą T 2 (T 2 < T 1) - lodówka, ilość ciepła przekazywanego na cyklQ 2
, podczas wykonywania pracy ALE =
Q 1
-
Q 2
cykl bezpośredni stosowane w silnikach cieplnych - okresowo pracujących silnikach, które wykonują pracę z powodu ciepła odbieranego z zewnątrz. Obieg odwrotny stosowany jest w urządzeniach chłodniczych – instalacjach pracujących okresowo, w których na skutek działania sił zewnętrznych ciepło jest przekazywane do ciała o wyższej temperaturze. W wyniku procesu okrężnego układ powraca do stanu pierwotnego, a zatem całkowita zmiana energii wewnętrznej wynosi zero. NastępnieІ
start t/d dla procesu okrężnego Q= Δ
u+
A=
A,
Oznacza to, że praca wykonana na cykl jest równa ilości ciepła otrzymanego z zewnątrz, ale Q=
Q 1 -
Q 2
Q 1 - ilość ciepło odbierane przez system, Q 2 - ilość ciepło wydzielane przez układ. Wydajność termiczna dla procesu kołowego jest równy stosunkowi pracy wykonanej przez układ do ilości ciepła dostarczonego do układu: Dla η = 1 warunekQ 2
= 0, tj. silnik cieplny musi mieć jedno źródło ciepłaQ 1
, ale jest to sprzeczne z drugą zasadą t/d. Proces odwrotny do tego, co dzieje się w silniku cieplnym, jest stosowany w maszynie chłodniczej. Q=
Q 2
-
Q 1
< 0, следовательно
A< 0.
Bez pracy nie można odebrać ciepła mniej nagrzanemu ciału i oddać go cieplejszemu. Na podstawie drugiego prawa t/d Carnot wydedukował twierdzenie. Twierdzenie Carnota: wszystkich okresowo pracujących silników cieplnych o tych samych temperaturach grzejnika ( T 1) i lodówki ( T 2), najwyższa wydajność. mają odwracalne maszyny. KPD odwracalne maszyny dla równych T 1 i T 2 są równe i nie zależą od rodzaju płynu roboczego. Ciało robocze to ciało, które wykonuje proces okrężny i wymienia energię z innymi ciałami. Cykl Carnota jest najbardziej ekonomicznym cyklem odwracalnym, składającym się z 2 izoterm i 2 adiabatów. 2-3 - adiabata. ekspansja, gaz działaA 2-3
>0 nad ciałami zewnętrznymi. 3-4 kompresja izotermiczna przy T 2 lodówki; odbierane jest ciepłoQ 2
i praca jest skończona Kompresja adiabatyczna 4-1, praca wykonywana jest na gazie 4-1 <0 внешними телами.
W procesie izotermicznymu= const , więc Q 1
=
A 12
Z ekspansją adiabatycznąQ 2-3 = 0, a praca z gazem A 23
wykonane energią wewnętrzną 23 = - u Ilość ciepłaQ 2
, oddane przez gaz do lodówki podczas kompresji izotermicznej jest równe pracy sprężania ALE 3-4
Praca kompresji adiabatycznej Praca wykonywana w procesie okrężnym A = A 12 + A 23 + A 34 + A 41 = Q 1 + A 23 - Q 2 - A 23 = Q 1 - Q 2
i jest równy polu krzywej 1-2-3-4-1. Wydajność termiczna Cykl Carnota Z równania adiabatycznego dla procesów 2-3 i 3-4 otrzymujemy Następnie tych. efektywność Cykl Carnota jest określony tylko przez temperatury grzejnika i chłodnicy. Aby zwiększyć wydajność trzeba zwiększyć różnicę T 1 - T 2 .
******************************************************* ******************************************************
W poprzedniej sekcji wyszliśmy z podstawowego założenia, że dla każdego systemu istnieje parametr zwany entropią i oznaczony przez S. Dla małych wartości interakcji termicznej odpowiednia różnicowa zmiana entropii dS wynosi . Używamy tej definicji poniżej do obliczania zmian entropii w niektórych prostych i dobrze znanych procesach. Zmiana entropii podczas topnienia lodu. Załóżmy, że w upalny letni dzień przynosimy na piknik termos wypełniony mieszanką lodu i wody. Ponieważ izolacja termosu nie jest idealna, lód będzie się stopniowo topił. Jednak topnienie następuje powoli, temperatura w termosie pozostanie prawie niezmieniona i równa 0 ° C. Obliczmy zmianę entropii odpowiadającą stopieniu 1 mola (lub 18 g) lodu. Wartość tabelaryczna ciepła topnienia lodu wynosi 79,67 cal/g, co daje około 1434 cal/mol. Wtedy można pisać Tak jak poprzednio, oznacza to po prostu sumowanie nieskończenie małych ilości - całkowanie (lub sumowanie) wszystkich wielkości odpowiadających każdej małej ilości ciepła. Całkowanie przeprowadza się w tym przypadku szczególnie po prostu dlatego, że temperatura T nie zmienia się podczas procesu topienia. Dlatego współczynnik 1/T można wyjąć spod znaku całki, tak aby stał się tylko czynnikiem przy ostatnim wyrażeniu to faktycznie ciepło przemiany fazowej (topnienia) lodu cal/mol. Zależność (19) oznacza, że entropia 1 mola wody w temperaturze 273 K jest o 5,27 cal/K wyższa niż entropia 1 mola lodu w tej samej temperaturze. Uwierz, kiedy lód się stopi. Entropia wzrośnie. I odwrotnie, jeśli z wody o temperaturze 273 K usunie się wystarczającą ilość ciepła, aby utworzyć 1 mol lodu o temperaturze 273 K, entropia układu zmniejszy się o . Zauważ, że w tej sekcji użyliśmy bezwzględnej temperatury Kelvina w mianowniku stosunku. Byłoby możliwe użycie bezwzględnej skali Rankina, jeśli mierzymy ilość ciepła w b.t. e. Jest oczywiste, że temperatury w stopniach Celsjusza lub Fahrenheita nie mogą być używane w mianowniku wyrażenia (co czasami próbują zrobić nawet przeszkoleni studenci). I tak np. posługując się skalą Celsjusza w rozpatrywanym przypadku doszlibyśmy do absurdalnego wyniku (mianownik wyrażenia zwróciłby się do zera). Zauważ, że jednostki, w których wyrażana jest zmiana entropii, pokrywają się z jednostkami, w których mierzona jest molowa pojemność cieplna Zmiana entropii, gdy 1 mol lodu topi się w temperaturze zamarzania w normalnych warunkach, wynosi 5,27 cal/(mol·K) . Zmiana entropii podczas gotowania wody. Innym dobrze znanym procesem zachodzącym w określonej temperaturze jest przemiana ciekłej wody w parę pod ciśnieniem 1 atm. Temperatura wrzenia wody w normalnych warunkach wynosi z definicji 100°C, czyli 373 K. Ciepło parowania w tej temperaturze wynosi 539 cal/g, czyli 9702 cal/mol. Wtedy zmiana entropii odpowiadająca odparowaniu 1 mola wody w normalnych warunkach wynosi To obliczenie okazało się o tyle proste, że temperatura nie zmieniała się w trakcie procesu. Należy zauważyć, że zmiana entropii w procesie parowania wody jest prawie 5 razy większa niż zmiana entropii w procesie topnienia lodu. Wartość jest nieco wyższa niż wartości typowe dla takich sytuacji i wskazuje na niezwykłe właściwości substancji, takiej jak woda. Dla wielu „normalnych” (niepolarnych) cieczy zmiana entropii podczas parowania wynosi. Reguła ta została uzyskana empirycznie przez angielskiego fizyka Fredericka Troutona (1863-1922) i nosi nazwę „reguły Troutona”. Pozwala oszacować ciepło parowania danej substancji, jeśli znana jest temperatura jej wrzenia w normalnych warunkach. Aby znaleźć przybliżoną wartość ciepła parowania, wystarczy pomnożyć temperaturę wrzenia (wyrażoną w kelwinach) przez stałą Growtona. Zmiana entropii podczas izotermicznej ekspansji gazu doskonałego. Jest jeszcze jeden proces w stałej temperaturze, z którym zetknęliśmy się już nie raz – jest to proces odwracalnej izotermicznej ekspansji gazu doskonałego. Jeśli wraz z termiką występuje tylko zwykłe oddziaływanie mechaniczne (tak, że elementarna praca jest wyrażona wzorem, pierwszą zasadę termodynamiki dla 1 mola gazu doskonałego można zapisać jako (tutaj bierze się to pod uwagę). Korzystając z równania pV = RT, dla dT = 0 (warunek stałej temperatury) możemy napisać Musieliśmy zintegrować to wyrażenie w rozdz. 4, więc oto wynik natychmiast: Ponieważ temperatura T pozostaje stała, wyrażenie na odpowiednią zmianę entropii ma postać Jak wiadomo, stała gazowa R ma wymiar cal/(mol K), a mnożnik zawierający logarytm jest liczbą bezwymiarową, tak że wymiary w lewej i prawej części relacji (24) pokrywają się. Zatem wzrostowi objętości (tj. rozszerzeniu) w stałej temperaturze towarzyszy wzrost entropii. Wróćmy do przypadku wrzącej wody. Niech odparuje 1 mol wody; 1 mol gazu doskonałego, jak pamiętamy, w normalnych warunkach (ciśnienie 1 atm i temperatura 273 K) zajmuje objętość około 22 400 cm3. Przy 373 K odpowiednia objętość wyniesie 22 400 (373/273), czyli około 30 600 cm3. Przed odparowaniem 1 mol cieczy zajmował objętość około, więc stosunek jest zgodny z równaniem (24), zmiana entropii odpowiadająca zmianie objętości w wyniku parowania wynosi R ln 1700. Biorąc pod uwagę, że wartość R wynosi w przybliżeniu równa , pożądana zmiana entropii wynosi około 14,88 cal/(mol K). Obliczając w poprzednim punkcie całkowitą zmianę entropii podczas całego procesu odparowania 1 mola wody, otrzymaliśmy wartość 26,0 cal/(mol K). Jak już widzieliśmy, nieco ponad połowa tej wartości jest związana ze zmianą objętości, gdy ciecz przechodzi w parę. Zmiany entropii spowodowane zmianami temperatury. Jak dotąd wszystkie nasze obliczenia zmiany entropii zostały przeprowadzone dla oddziaływań termicznych w stałej temperaturze. Rozważmy teraz bardziej powszechny i nieco bardziej skomplikowany przypadek, gdy odwracalne ogrzewanie prowadzi do zmiany temperatury. Jeśli ogrzewanie zachodzi przy stałej objętości, to. zgodnie z definicją ciepła właściwego przy stałej objętości mamy . Następnie Całkując to wyrażenie w skończonym zakresie temperatur, otrzymujemy Przyjęto tutaj, że pojemność cieplna nie zależy od temperatury i można ją wyjąć ze znaku całki. Znaczące jest to, że identyfikując usuwamy ograniczenie odwracalności procesu nagrzewania, a także równomierności temperatury podczas procesu nagrzewania. Temperaturę układu musimy znać tylko na początku i na końcu procesu grzania. Innymi słowy, równowaga termiczna musi istnieć tylko w stanie początkowym i końcowym: stany pośrednie nie odgrywają żadnej roli. W bardziej powszechnym i praktycznie znacznie łatwiejszym przypadku ogrzewania przy stałym ciśnieniu mamy . Dosłownie powtarzając wszystkie powyższe rozumowania, otrzymujemy 2. Ogrzewanie wody przy 1 atm od 273 K do 373 K: 3. Przemiana wodno-parowa przy 1 atm i 373 K: Zatem wynikająca z tego zmiana entropii podczas przemiany 1 mola lodu o temperaturze 273 K w parę o temperaturze 373 K wynosi Druga zasada termodynamiki określa kryteria nieodwracalności procesów termodynamicznych. Istnieje wiele sformułowań drugiego prawa, które są sobie równoważne. Przedstawiamy tutaj tylko jedno sformułowanie związane z entropią. istnieje funkcja stanu- entropia S, który ma następującą właściwość: , (4.1) gdzie znak równości odnosi się do procesów odwracalnych, a znak większego niż do procesów nieodwracalnych. W przypadku systemów izolowanych drugie prawo stanowi: dS i 0, (4.2) tj. entropia układów izolowanych w procesach nieodwracalnych może tylko wzrastać, aw stanie równowagi termodynamicznej osiąga maksimum ( dS = 0, Nazywa się nierówność (4.1). Nierówność Clausiusa. Ponieważ entropia jest funkcją stanu, jej zmiana w dowolnym procesie cyklicznym wynosi 0, dlatego dla procesów cyklicznych nierówność Clausiusa ma postać: gdzie stawia się znak równości, jeśli cały cykl jest całkowicie odwracalny. Entropię można określić za pomocą dwóch równoważnych podejść - statystycznego i termodynamicznego. Definicja statystyczna opiera się na założeniu, że nieodwracalne procesy w termodynamice są spowodowane przejściem do stanu bardziej prawdopodobnego, więc entropię można powiązać z prawdopodobieństwem: gdzie k= 1,38 10 -23 J/K - stała Boltzmanna (k = R / N A) W- tzw. prawdopodobieństwo termodynamiczne, tj. liczba mikrostanów, które odpowiadają danemu makrostanowi systemu (patrz rozdział 10). Nazywa się formuła (4.4). Formuła Boltzmanna. Z punktu widzenia ścisłej termodynamiki statystycznej entropię wprowadza się następująco: gdzie G( mi) to objętość fazowa zajmowana przez zespół mikrokanoniczny z energią mi. Definicja termodynamiczna entropia opiera się na uwzględnieniu procesów odwracalnych: Ta definicja pozwala nam przedstawić ciepło elementarne w tej samej formie, co różne rodzaje pracy: Q arr = TdS, (4.7) gdzie temperatura odgrywa rolę siły uogólnionej, a entropia rolę uogólnionej (termicznej) współrzędnej. Obliczanie zmiany entropii dla różnych procesów Obliczenia termodynamiczne zmiany entropii opierają się na definicji (4.6) oraz na własnościach pochodnych cząstkowych entropii względem parametrów termodynamicznych: Dwie ostatnie tożsamości są Relacje Maxwella(patrz wyprowadzenie w rozdziale 5). 1) Ogrzewanie lub chłodzenie przy stałym ciśnieniu. Ilość ciepła potrzebną do zmiany temperatury układu wyraża się za pomocą pojemności cieplnej: Q arr = Cp dT. Jeśli pojemność cieplna nie zależy od temperatury w przedziale od T 1 do T 2 , to równanie (4.8) można scałkować: Jeżeli zmiana temperatury zachodzi przy stałej objętości, to we wzorach (4.9) i (4.10) Cp należy wymienić na C V. 2) Izotermiczne rozszerzanie lub kurczenie się. Aby obliczyć entropię w tym przypadku, musisz znać równanie stanu układu. Obliczenia opierają się na wykorzystaniu zależności Maxwella: W szczególności dla izotermicznej ekspansji gazu doskonałego ( p = nRT / V) Ten sam wynik można uzyskać za pomocą wyrażenia na ciepło izotermicznej odwracalnej ekspansji gazu doskonałego: Q arr = nRT ln( V 2 /V 1) .
3) Przejścia fazowe. Przy odwracalnym przejściu fazowym temperatura pozostaje stała, a ciepło przemiany fazowej przy stałym ciśnieniu jest H fp, więc zmiana entropii wynosi: Podczas topienia i wrzenia ciepło jest pochłaniane, więc entropia w tych procesach wzrasta: S telewizja< S oraz< S d. W tym przypadku entropia środowiska zmniejsza się o wartość S f.p. , więc zmiana entropii Wszechświata wynosi 0, zgodnie z oczekiwaniami dla procesu odwracalnego w układzie izolowanym. 4) Mieszanie gazów doskonałych w stałej temperaturze i ciśnieniu. Jeśli n 1 mol jednego gazu zajmuje objętość V 1, zmieszany z n 2 mole innego gazu zajmują objętość V 2 , wtedy całkowita objętość będzie równa V 1 + V 2 , a gazy rozszerzają się niezależnie od siebie, a całkowita zmiana entropii jest równa sumie zmian entropii każdego gazu: gdzie x ja- ułamek molowy i gaz w powstałej mieszaninie gazowej. Zmiana entropii (4.14) jest zawsze dodatnia, ponieważ wszystkie ln x ja < 0,
поэтому идеальные газы всегда смешиваются
необратимо. Jeżeli w tych samych warunkach zmieszane zostaną dwie porcje tego samego gazu, to równanie (4.14) nie ma już zastosowania. Nie ma żadnych zmian w systemie podczas mieszania i S= 0. Niemniej jednak wzór (4.14) nie zawiera żadnych indywidualnych parametrów gazów, dlatego wydaje się, że powinien mieć zastosowanie do mieszania identycznych gazów. Ta sprzeczność nazywa się Paradoks Gibbsa. Absolutna entropia W przeciwieństwie do wielu innych funkcji termodynamicznych, entropia ma punkt odniesienia, który jest określony przez Postulat Plancka (trzecia zasada termodynamiki): Przy zera absolutnym T= 0 K wszystkie idealne kryształy Ponieważ temperatura dąży do zera absolutnego, nie tylko entropia dąży do 0, ale także jej pochodne w odniesieniu do wszystkich parametrów termodynamicznych: (x = p, V).
(4.15) Oznacza to, że w pobliżu zera absolutnego wszystkie procesy termodynamiczne przebiegają bez zmiany entropii. To stwierdzenie nazywa się termiczne twierdzenie Nernsta. Postulat Plancka pozwala nam wprowadzić to pojęcie absolutna entropia substancje, tj. entropia liczona od zera w godz T= 0. Aby obliczyć bezwzględną entropię substancji w stanie standardowym, konieczna jest znajomość zależności pojemności cieplnej Cp od temperatury dla każdej z faz, a także od temperatury i entalpii przejść fazowych. Na przykład entropia bezwzględna substancji gazowej w stanie standardowym w temperaturze T składa się z następujących elementów: Tabele termodynamiczne zwykle podają bezwzględne wartości entropii w stanie standardowym w temperaturze 298 K. Wartości bezwzględnej entropii substancji służą do obliczania zmiany entropii w reakcjach chemicznych: PRZYKŁADY Przykład 4-1. Wyznacz zależność entropii od objętości dla układu termodynamicznego opisanego równaniem stanu (dla jednego mola) Rozwiązanie. Całkując tę równość, znajdujemy zależność entropii od objętości: gdzie konst zależne od temperatury. Przykład 4-2. Oblicz zmianę entropii po podgrzaniu 0,7 mola siarki jednoskośnej od temperatury 25 do 200 o C pod ciśnieniem 1 atm. Molowa pojemność cieplna siarki wynosi: C p (S tv) \u003d 23,64 J / (mol. K), Temperatura topnienia siarki jednoskośnej wynosi 119 o C, ciepło topnienia 45,2 J/g. Rozwiązanie. Całkowita zmiana entropii składa się z trzech składowych: 1) podgrzania siarki stałej od 25 do 119°C, 2) stopienia, 3) podgrzania siarki płynnej od 119 do 200°C. S = S 1
+ S 2 + S 3 = 11,88 J/K. Odpowiadać. 11,88 J/K. Przykład 4-3. Znajdź zmianę entropii gazu i otoczenia, jeśli n mole gazu doskonałego rozszerzają się izotermicznie z objętością V 1 do głośności V p. Rozwiązanie. a) Zmianę entropii gazu podczas odwracalnego rozprężania izotermicznego można znaleźć, korzystając z termodynamicznej definicji entropii z obliczeniem ciepła rozprężania zgodnie z pierwszą zasadą: Ponieważ ekspansja jest odwracalna, całkowita zmiana entropii Wszechświata wynosi 0, więc zmiana entropii otoczenia jest równa zmianie entropii gazu o przeciwnym znaku: b) Entropia jest funkcją stanu, więc zmiana entropii układu nie zależy od tego, jak przebiegał proces – odwracalny czy nieodwracalny. Zmiana entropii gazu podczas nieodwracalnego rozprężania pod ciśnieniem zewnętrznym będzie taka sama jak podczas odwracalnego rozprężania. Kolejną rzeczą jest entropia otoczenia, którą można znaleźć, obliczając, korzystając z pierwszego prawa, ciepło przekazane do układu: W tym wyprowadzeniu wykorzystaliśmy fakt, że u= 0 (temperatura jest stała). Praca wykonana przez układ przy stałym ciśnieniu zewnętrznym jest równa: A = p(V 2 -V 1), a ciepło odebrane przez otoczenie jest równe pracy wykonanej przez układ, ze znakiem przeciwnym. Całkowita zmiana entropii gazu i otoczenia jest większa od 0: zgodnie z oczekiwaniami dla procesu nieodwracalnego. Przykład 4-4. Oblicz zmianę entropii 1000 g wody w wyniku jej zamarznięcia w temperaturze -5°C. Ciepło topnienia lodu w temperaturze 0°C wynosi 6008 J/mol. Pojemności cieplne lodu i wody wynoszą odpowiednio 34,7 i 75,3 J/(mol·K). Wyjaśnij, dlaczego entropia maleje podczas zamrażania, chociaż proces ten jest spontaniczny. Rozwiązanie. Nieodwracalny proces zamrażania wody w temperaturze -5 ° C można przedstawić jako sekwencję procesów odwracalnych: 1) podgrzewanie wody z Zmianę entropii w pierwszym i trzecim procesie (podczas zmiany temperatury) oblicza się ze wzoru (4.9): Zmianę entropii w drugim procesie oblicza się jak dla zwykłego przejścia fazowego (4.13). Należy tylko pamiętać, że podczas zamrażania uwalniane jest ciepło: Dlatego entropia jest funkcją stanu, całkowita zmiana entropii jest równa sumie tych trzech procesów: S = S 1
+ S 2 + S 3 = -1181 J/K. Entropia spada podczas zamrażania, chociaż proces jest spontaniczny. Wynika to z faktu, że ciepło jest uwalniane do otoczenia i entropia środowiska wzrasta, a wzrost ten jest większy niż 1181 J / K, więc entropia Wszechświata wzrasta, gdy woda zamarza, zgodnie z oczekiwaniami w procesie nieodwracalnym. Odpowiadać. -1181 J/K. ZADANIA 4-1. Podaj przykład procesu termodynamicznego, który można przeprowadzić zarówno odwracalnie, jak i nieodwracalnie. Oblicz zmianę entropii układu i środowiska w obu przypadkach. 4-2. Sprawdź nierówność Clausiusa dla procesu cyklicznego przedstawionego w zadaniu 2.14. 4-3. Oblicz entropię molową neonu w temperaturze 500 K, jeśli w temperaturze 298 K i tej samej objętości entropia neonu wynosi 146,2 J/(mol·K). 4-4. Oblicz zmianę entropii, gdy 11,2 litra azotu zostanie ogrzane od 0 do 50 o C i jednocześnie zmniejszy się ciśnienie z 1 atm do 0,01 atm. 4-5. Jeden mol helu w temperaturze 100 o C i pod ciśnieniem 1 atm miesza się z 0,5 mola neonu w temperaturze 0 o C i pod ciśnieniem 1 atm. Wyznacz zmianę entropii, jeśli ciśnienie końcowe wynosi 1 atm. 4-6. Oblicz zmianę entropii podczas tworzenia 1 m3 powietrza z azotu i tlenu (20% obj.) w temperaturze 25 ° C i pod ciśnieniem 1 atm. 4-7. Trzy mole doskonałego jednoatomowego gazu ( C V = 3,0 cal / (mol. K)), znajduje się w T 1 = 350 K i P 1 = 5,0 atm, odwracalnie i adiabatycznie rozszerza się do ciśnienia P 2 = 1,0 atm. Oblicz końcową temperaturę i objętość, a także wykonaną pracę i zmianę energii wewnętrznej, entalpii i entropii w tym procesie. 4-8. Oblicz zmianę entropii po podgrzaniu 0,4 mola chlorku sodu z 20 do 850 ° C. Molowa pojemność cieplna chlorku sodu wynosi: C p (NaCl tv) = 45,94 + 16,32. 10 -3 . T J / (mol. K), Temperatura topnienia chlorku sodu wynosi 800 o C, ciepło topnienia wynosi 31,0 kJ/mol. 4-9. Oblicz zmianę entropii podczas mieszania 5 kg wody o temperaturze 80 ° C z 10 kg wody o temperaturze 20 ° C. Weź ciepło właściwe wody równe: Cp(H2O) = 4,184 J/(g. K). 4-10. Oblicz zmianę entropii po dodaniu 200 g lodu o temperaturze 0°C do 200 g wody (90°C) w izolowanym naczyniu. Ciepło topnienia lodu wynosi 6,0 kJ/mol. 4-11. Dla pewnego ciała stałego zależność współczynnika rozszerzalności od ciśnienia znajduje się w zakresie ciśnień od p 1 do p 2: O ile zmniejszy się entropia tego ciała po ściśnięciu p 1 do p 2 ? 4-12. Znajdź zmianę entropii gazu i otoczenia, jeśli n mole gazu doskonałego rozszerzają się izotermicznie pod wpływem ciśnienia p 1 do ciśnienia p 2: a) odwracalny; b) przed naciskiem zewnętrznym p < p 2 . 4-13. Zapisz wzór na obliczenie entropii bezwzględnej jednego mola wody w temperaturze 300 0 C i pod ciśnieniem 2 atm. 4-14. Narysuj wykres standardowej entropii wody w funkcji temperatury w zakresie od 0 do 400 K. 4-15. Zapisz entropię jednego mola gazu doskonałego w funkcji temperatury i ciśnienia (uwzględniając stałą pojemności cieplnej). 4-16. Wyznacz zależność entropii od objętości dla układu termodynamicznego opisanego równaniem stanu (dla jednego mola): 4-17. Wyznacz zależność entropii od objętości dla układu termodynamicznego opisanego równaniem stanu (dla jednego mola): 4-18. Jeden mol gazu jest opisany równaniem stanu gdzie f(V) to funkcja niezależna od temperatury. Oblicz zmianę entropii gazu podczas jego nieodwracalnego izotermicznego rozszerzania się wraz z objętością V 1 do głośności V 2 . 4-19. Oblicz zmianę entropii 1000 g metanolu w wyniku jego zamrożenia w temperaturze -105°C. Ciepło topnienia stałego metanolu w temperaturze -98°C (tt.) wynosi 3160 J/mol. Pojemności cieplne stałego i ciekłego metanolu wynoszą odpowiednio 55,6 i 81,6 J/(mol·K). Wyjaśnij, dlaczego entropia maleje podczas zamrażania, chociaż proces ten jest spontaniczny. 4-20. Pojemność cieplna jakiejś substancji w zakresie temperatur od T 1 do T 2 zmienia się w następujący sposób: Narysuj zależność entropii substancji od temperatury w tym zakresie temperatur. 4-21. Korzystając z danych odniesienia, podaj przykład spontanicznej reakcji chemicznej, dla której standardowa zmiana entropii jest mniejsza od 0. 4-22. Korzystając z danych referencyjnych, oblicz standardową zmianę entropii w reakcji H 2 (g) + SO 2 (g) \u003d H 2 O (g) a) w 25 ° C; b) w temperaturze 300 o C.
.
(147)

![]()
![]()
![]()
![]()
![]()
![]() ). Zatem entropia jest określana przez logarytm liczby stanów, w których można zrealizować dany mikrostan. Entropię można traktować jako miarę prawdopodobieństwa stanu układu t/d. Formuła Boltzmanna pozwala nam nadać entropii następującą interpretację statystyczną. Entropia jest miarą nieuporządkowania układu. Rzeczywiście, im większa liczba mikrostanów realizujących dany mikrostan, tym większa entropia. W stanie równowagi układu – najbardziej prawdopodobnym stanie układu – liczba mikrostanów jest maksymalna, entropia też jest maksymalna.
). Zatem entropia jest określana przez logarytm liczby stanów, w których można zrealizować dany mikrostan. Entropię można traktować jako miarę prawdopodobieństwa stanu układu t/d. Formuła Boltzmanna pozwala nam nadać entropii następującą interpretację statystyczną. Entropia jest miarą nieuporządkowania układu. Rzeczywiście, im większa liczba mikrostanów realizujących dany mikrostan, tym większa entropia. W stanie równowagi układu – najbardziej prawdopodobnym stanie układu – liczba mikrostanów jest maksymalna, entropia też jest maksymalna. Termostat- jest to układ t/d, który może wymieniać ciepło z ciałami bez zmiany temperatury.
Termostat- jest to układ t/d, który może wymieniać ciepło z ciałami bez zmiany temperatury. Okrągły proces lub cykl to proces, w którym system po przejściu przez szereg stanów powraca do stanu pierwotnego. Na diagramie stanu cykl jest reprezentowany przez zamkniętą krzywą. Cykl wykonywany przez gaz doskonały można podzielić na procesy rozprężania (1-2) i sprężania (2-1), praca rozprężania jest dodatnia ALE 1-2 > 0, ponieważV 2
>
V 1
, praca kompresji jest ujemna ALE 1-2 < 0, т.к.
V 2
<
V 1
. Dlatego praca wykonywana przez gaz na cykl jest określona przez pole powierzchni objęte zamkniętą krzywą 1-2-1. Jeśli praca dodatnia jest wykonywana w cyklu (cykl jest zgodny z ruchem wskazówek zegara), wówczas cykl nazywa się prostym, jeśli jest to cykl odwrotny (cykl odbywa się w kierunku przeciwnym do ruchu wskazówek zegara).
Okrągły proces lub cykl to proces, w którym system po przejściu przez szereg stanów powraca do stanu pierwotnego. Na diagramie stanu cykl jest reprezentowany przez zamkniętą krzywą. Cykl wykonywany przez gaz doskonały można podzielić na procesy rozprężania (1-2) i sprężania (2-1), praca rozprężania jest dodatnia ALE 1-2 > 0, ponieważV 2
>
V 1
, praca kompresji jest ujemna ALE 1-2 < 0, т.к.
V 2
<
V 1
. Dlatego praca wykonywana przez gaz na cykl jest określona przez pole powierzchni objęte zamkniętą krzywą 1-2-1. Jeśli praca dodatnia jest wykonywana w cyklu (cykl jest zgodny z ruchem wskazówek zegara), wówczas cykl nazywa się prostym, jeśli jest to cykl odwrotny (cykl odbywa się w kierunku przeciwnym do ruchu wskazówek zegara).![]()
 Z termostatu z temperaturą T 2 ilość ciepła jest odbieranaQ 2
i przesyłane do termostatu z temperaturąT 1
, ilość ciepłaQ 1
.
Z termostatu z temperaturą T 2 ilość ciepła jest odbieranaQ 2
i przesyłane do termostatu z temperaturąT 1
, ilość ciepłaQ 1
.
 1-2-rozprężanie izotermiczne przy T 1 grzałka; ciepło jest dostarczane do gazuQ 1
i praca jest skończona
1-2-rozprężanie izotermiczne przy T 1 grzałka; ciepło jest dostarczane do gazuQ 1
i praca jest skończona![]()
![]() ;
;
![]() 1
1
![]()
![]() 2
2
![]()
![]()
![]()
![]()



![]()

d 2 S < 0). (4.8)
(4.8)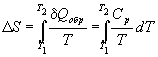 (4.9)
(4.9)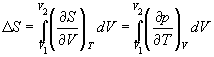 (4.11)
(4.11)![]() (4.13)
(4.13)
mają tę samą entropię równą zeru.![]() . (4.17)
. (4.17)![]()
![]()
Cp(Sw) \u003d 35,73 + 1,17. 10 -3 . T J/(mol. K). 4,54 J/K.
4,54 J/K.![]() 2,58 J/K.
2,58 J/K.
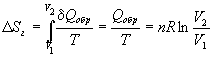 .
.![]() .
.![]() .
.![]() ,
,
-5 o C do punktu zamarzania (0 o C); 2) zamarzanie wody w temperaturze 0°C; 3) chłodzenie lodem od 0 do -5 °C:
 77,3 J/K.
77,3 J/K. -35,6 J/K.
-35,6 J/K.![]() -1223 J/K.
-1223 J/K.
Cp(NaClw) = 66,53 J/(mol·K).![]() .
.![]()
![]()